I will write a five-part series tutorial on implementing the python code to compute different sets of 2D molecular descriptors & fingerprints which are highly used in the context of drug discovery. Many thanks to the first-year Ph.D. students who request me to write tutorials on cheminformatics topics such as these. I welcome readers to request tutorial topics as well.
First, let’s get to know a bit of the background on molecular descriptors and fingerprints.
What are molecular descriptors or fingerprints?
A molecular descriptor is formally defined as:
“The final result of a logical and mathematical procedure, which transforms chemical information encoded within a symbolic representation of a molecule into a useful number or the result of some standardized experiment.”
Todeschini R, Consonni V (2009) Molecular descriptors for chemoinformatics. Wiley-VCH, Weinheim
A chemical structure can be characterized by a set of numerical values known as molecular fingerprints or descriptors. They may present properties of molecules such as log P, molecular weight, hydrogen bond donors, acceptors, rotatable bonds, etc. that can be linked to experimental evidence on the molecule. Or they may be 2D Fragment-based fingerprints presented by bit arrays of 0s and 1s wherein each bit position indicates the presence or absence of structural fragments (see Figure 1), e.g. MACCS (166 bits).

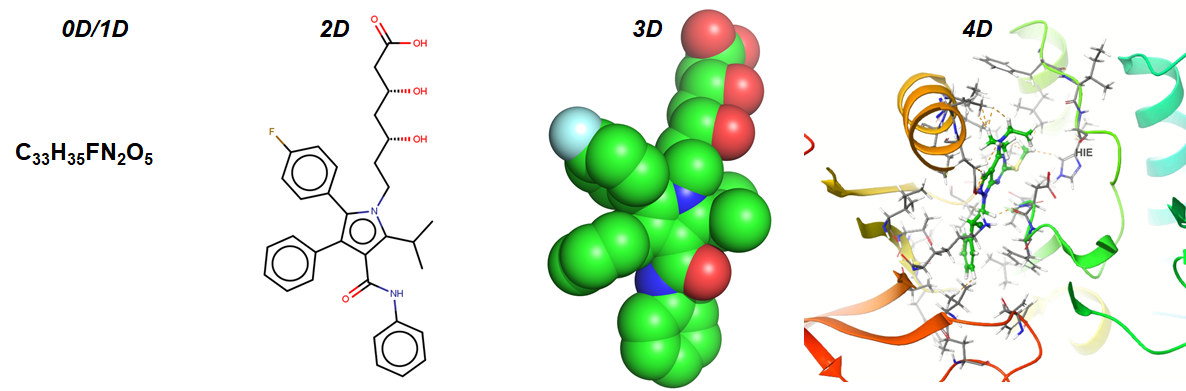
For each level of molecular representation (see Figure 2) such as 1D, 2D, 3D, or even 4D/MD (molecular dynamics time series based dimension), we can compute hundreds or thousands of structural features. There is a wide variety of molecular descriptors and fingerprints encoding constitutional, topological, geometrical, electrostatic, quantum-chemical, thermodynamical, fragmental features, and so on.

0-Dimensional (0D)
The representation of a molecule can be as simple as the chemical formula such as C33H35FN2O5, which indicates the presence of 33 Carbon atoms, 35 Hydrogen atoms, 1 Fluorine atom, 2 Nitrogen atoms and 5 Oxygen atoms. 0D representation does not convey much information about the molecular structure and atom connectivity. Some examples of 0D/1D descriptors are atom counts, bond counts, molecular weight, molar refractivity, etc.
1-Dimensional (1D)
1D descriptors usually account for fragment counts, hydrogen bond donors, acceptors, PSA, etc. They are usually binary values which indicate the presence or absence of given substructures or occurrence frequencies.
2-Dimensional (2D)
2D representation accounts for topo-structural features and how the atoms are connected (e.g., adjacency, connectivity). They are usually the descriptors derived the molecular graph representations of nodes and edges such as size, degree of branching, the neighborhood of atoms in terms of electronic & steric effects, flexibility, overall shape, etc.
3-Dimensional (3D)
In 3D, a molecule is viewed in space with x, y, z coordinates, and thus 3D derived descriptors can have high content information which can be beneficial for modeling biological endpoints of interest. 3D representation accounts for spatial and geometrical configuration, shape-based information, conformation-dependent distances, and surface properties (such as Van der Waals, Solvent-Accessible surface area). However, it can have several complications when it comes to the choice of the optimization method, and the resulting conformational arrangement, highly flexible molecules with multiple favorable conformers, the difference between experimental and computationally optimized conformations. It can also be time-consuming to perform the process of generating 3D conformers, assessing, and finalizing the optimized conformation and computing 3D descriptors. To note, I have seen in several cheminformatics projects that 2D descriptors generally perform as well as 3D descriptors in QSAR modeling, and also, 2D descriptors are very fast to compute.
4-Dimensional (4D)
4D descriptors are usually derived from reference grids and molecular dynamic simulations. Here, we quantitatively detect and characterize the interactions between a ligand and the amino acid residues in a receptor’s active site. This can be done by applying grid-based QSAR techniques wherein a molecule is placed in a 3D grid of evenly spaced grid points and computationally simulated and probed to estimate the surface of the molecule and possible protein-ligand interactions that can form (e.g. pi-pi stacking, hydrophobic, etc.). There are also alignment-dependent and ligand-based methods such as Comparative Molecular Field Analysis [CoMFA] and Comparative Molecular Similarity Indices Analysis [CoMSIA].
The molecular descriptors have many useful applications in drug discovery; one major application being the predictive machine learning model development, also known as QSAR (quantitative structure-activity relationship) modeling. You will see other uses of molecular descriptors in the other blog posts that I have written or will write in the future. For now, we will focus on how to write code in python for computing four sets of molecular descriptors.
Overview of the python tutorial
I have put together four python classes on 2D descriptor sets that can be imported and applied easily. These python classes are as follows and can be found in the Github repository:
- MACCS (part – 2)
- ECFP6 (part – 3)
- RDKit_2D (part – 4)
- Mordred_MRC_Descriptors (part – 5)
We call these python classes from the main python file (named as main.py). By running main.py script, you will compute all the aforementioned descriptors and generate them as separate CSV files. So, in the end, you will have the output files as shown in Figure 3 below.

If you want to learn more about “classes” in python, please check it out here. Stay tuned for the next blog post, MACCS Fingerprints in Python – Part 2 where I will explain in-depth about the code.