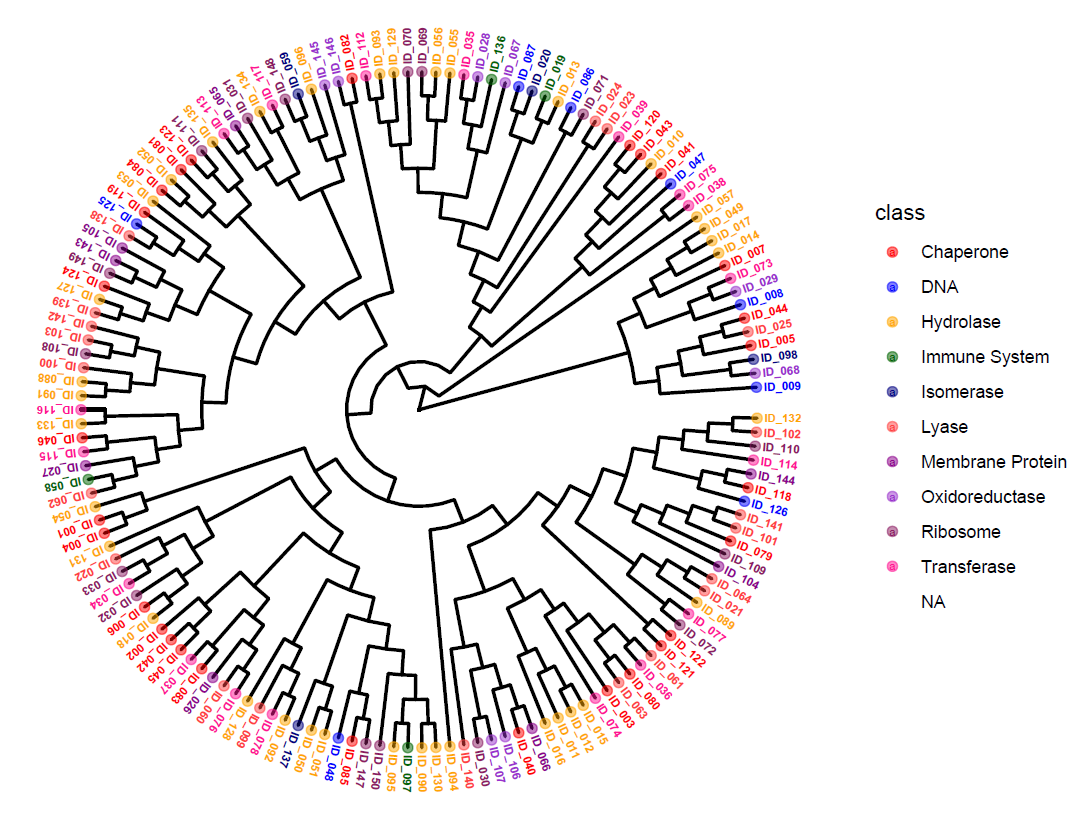
Today’s tutorial is on applying unsupervised hierarchical clustering in R and generating circular dendrograms with nodes colored based on discrete categories, like in the figure shown below (Figure 1).

Disclaimer: The above figure is generated with fake chemical data taken from different projects already published from my PhD years.
I used R 4.0.2 and R studio 1.3.959 and these packages: “ape”, “ggtree“, “phangorn“, “ggplot2“, so make sure you have them downloaded and installed. Most of the time, you can conveniently install packages that you want in R by typing install.packages(‘name-of-package’). But it doesn’t work in certain scenarios so you have to go the official website and follow their instructions.
I have often used unsupervised hierarchical clustering and explained it in my previous posts so I won’t go into the details here. The entire code is also available in the repository in my GitHub account.
Alright, let’s take a quick glance at the CSV file (Figure 2) that we will be using, and then let’s get down to the codes.

Each row is a chemical structure which contains the ID (used to label the nodes), Class (used to color the nodes so we know which class each chemical is annotated with), and chemical descriptors (which are used in constructing the unsupervised hierarchical structure of the dendrogram).
First, let’s make sure we have the required packages properly installed by running the code section below. It will print out TRUE for each of the package installed.
list.of.packages <- c("ape","ggtree","phangorn","ggplot2")
new.packages <- list.of.packages[!(list.of.packages %in% installed.packages()[,"Package"])]
if(length(new.packages)) install.packages(new.packages)
lapply(list.of.packages, require, character.only = TRUE)
rm(list=ls())
Next, let’s read the csv file and set the row names.
csv_data <- read.csv('sample.csv',header=TRUE,sep=",")
rownames(csv_data) <- csv_data[,1]
Note that, we set the very first column (csv_data[,1] )which contain the IDs of chemicals (ID_001, ID_002, ID_003, etc.) as the row names. Only when you set the row names, the nodes will be labeled properly when you generate the dendrograms.
Then, we will subset the data that contains the descriptors only. In the sample CSV file, all the columns contain descriptors except for ‘ID” and ‘Class”. So, we set the variable desc to contain all the other columns except for these two.
myvars <- names(csv_data) %in% c('ID', 'Class')
desc <- csv_data[!myvars]
We want to color the nodes based on different categories in “Class” column. So, we will set up a variable called “class_data” that combines “Class” column to “desc” data frame.
class <- csv_data$Class
class_data <- cbind(as.data.frame(row.names(desc)),as.data.frame(class))
We will then scale the columns of the “desc” data frame since it is a non-binary data. Scaling the data is a standard step in the clustering procedure when you work with continuous data. We don’t need to apply the “scaling” step when we work with bit-based or binary data such as MACCS or ECFP fingerprints.
matTrans <- scale(desc)
Then, we compute the distance matrix on the scaled data frame by applying dist() function with default Euclidean distance method and perform unsupervised hierarchical clustering using the Ward’s minimum variance method.
d <- dist(matTrans)
tupgma <- upgma(d, method="ward.D2")
Okay, we now have almost everything ready to actually get to the visualization part. But we still need to do one more step. We need to set a color palette for the different categories in “Class” column. In order to find out how many categories are there, we can run the following command:
unique(csv_data$Class)
It will print out the following. So, in total, we have 10 different categories. Note the way R console numbers the unique values for our convenience in counting especially in the last line.
[1] “Chaperone” “DNA”
[3] “Hydrolase” “Immune System”
[5] “Isomerase” “Lyase”
[7] “Membrane Protein” “Oxidoreductase”
[9] “Ribosome” “Transferase”
We will manually handpick the colors that we would like for each category and store them in the variable “cols“. You can check out the colors in R document here for more colors that may be of interest to you. Make sure you have the right number of colors selected for the number of categories you have.
cols <- c("red", "blue1", "orange", "darkgreen", "navy", "brown1", "darkmagenta","darkorchid", "maroon4", "deeppink1")
At this point, we have pretty much all we need to generate the visualizations. We will call the ggtree package and set the layout to “circular” since we are going for circular dendrograms. You can choose other layouts based on your preference by checking out this blog post here.
You can also look up the documentation and adjust the parameters accordingly that will work well for your visualization. I think many are self-explanatory so I won’t go into each parameter used in the script here.
# Adjust the parameters accordingly.
t4 <- ggtree(tupgma, layout="circular", size = 1)
t4 <- t4 %<+% class_data +
geom_text(aes(color = class, label = label, angle = angle,
hjust = - 0.2, fontface = "bold"), size = 2.6) +
geom_tippoint(aes(color = class), alpha = 0.5, size = 2) +
scale_color_manual(values = cols) +
theme(legend.position="right") +
theme(plot.margin = unit(c(0,0,0,0), "cm")) +
theme(plot.background = element_rect(fill = 'white', colour = 'white')) +
theme(panel.background = element_rect(fill = 'white', colour = 'white')) +
theme(legend.background = element_rect(fill = 'white'),
legend.title = element_text(size = 8,face = "bold")) + # LEGEND TITLE
theme(legend.text = element_text(size = 8, face = "bold")) + # LEGEND TEXT
theme(text = element_text(color = "black", size = 8)) +
geom_treescale(x = 0.1,y = 0.1, width = 0.1, offset = NULL,
color = "white", linesize = 1E-100, fontsize = 1E-100)
print(t4)
In the end, you should have visualization as shown in Figure 3. You can see that the nodes are colored based on their class of Chaperone, DNA, Hydrolase, etc. and you also have the legend with the colors and classes clarified.
We can set branch length to None to visualize the dendrogram without branch length scaling and only display the tree structure, and we will have the visualization as in Figure 4.

For example, if you use the default layout with the code snippet below, you would have the following figure (Figure 5).
t4 <- ggtree(tupgma, size = 0.7)
t4 <- t4 %<+% class_data +
geom_text(aes(color = class, label = label, angle = 0,
hjust = - 0.2, fontface = "bold"), size = 1.3) +
geom_tippoint(aes(color = class), alpha = 0.5, size = 1.3) +
scale_color_manual(values = cols)
print(t4)

Let’s try now with “slanted” layout to see a different visualization (Figure 6).
# Adjust the parameters accordingly.
t4 <- ggtree(tupgma, layout="slanted", size = 0.7)
t4 <- t4 %<+% class_data +
geom_text(aes(color = class, label = label, angle = 0,
hjust = - 0.2, fontface = "bold"), size = 1.3) +
geom_tippoint(aes(color = class), alpha = 0.5, size = 1.3) +
scale_color_manual(values = cols)
print(t4)

There is a way to tweak the parameters in ggtree or change the theme to have a black version of your visualization. You can also use an external photo editing software to achieve a different look like in FIgure 7.

I hope it has been helpful!
I will also make another post where you can color the nodes of the dendrogram on a continuous scale. Please let me know if you have any feedbacks, suggestions and if you find any mistakes. Thank you!