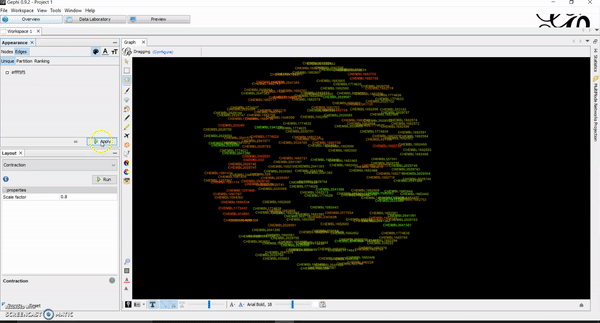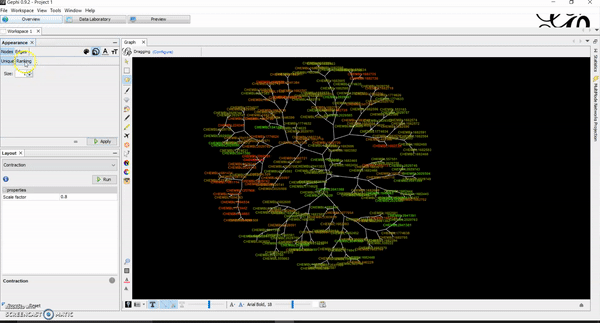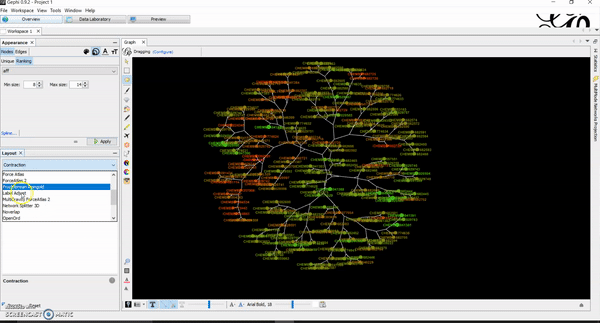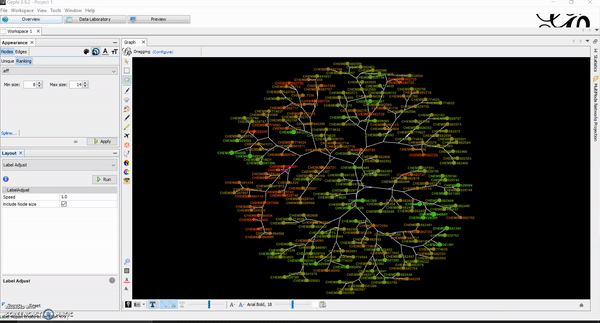
Today, I want to write a tutorial on how to generate chemical space visualizations using a combination of R and Gephi. I have found them to be a powerful way of assessing the chemical data and finding hidden patterns that could be crucial in estimating the biological endpoints of interest. Before we go on, let me hook you with an end result similar to what you will be creating at the end of this tutorial.

Chemical space visualization of MacrolactoneDB using ECFP6 chemical descriptors, Euclidean distance, and Ward’s linkage. Each node is a chemical structure colored by its experimental value in the form of pChEMBL.First and foremost, you will need to download Gephi visualization tool, and R statistical language (I use Rstudio for the IDE).
We will use a sample chemical dataset provided in this github repository This file contains ChEMBL IDs, 2D RDKit descriptors generated using open-sourced library RDKit in python, and “Aff” values (negative log of IC50s).
In generating gephi networks, you will need two input files:
- Nodes
- Edges
There are other variations like inputting adjacency matrix, but we will do it this way in this tutorial.
First, we will perform unsupervised hierarchical clustering and generate network data (i.e. nodes and edges) using these chemical descriptors in R. This method will allow us to group molecules with similar chemical properties and/or structural fragments (depending on what type of chemical descriptors you use). From this, we will generate the edge file required for gephi networks.
The R code (along with comments) associated with conducting hierarchical clustering and generating edges has been provided in gephi_network.R.
### The code snippet below will allow you to install the required library “phangorn” if it hasn’t already been installed. ###
list.of.packages <- c("phangorn")
new.packages <- list.of.packages[!(list.of.packages %in% installed.packages()[,"Package"])]
if(length(new.packages)) install.packages(new.packages)
lapply(list.of.packages, require, character.only = TRUE)
rm(list=ls())### Then, we will load the file containing ChEMBL IDs, 2D rdkit descriptors and “Aff” values. ###
csv_data <- read.csv('2Drdkit_chembl364.csv',header=TRUE,sep=",")### We will subset the data so that we are clustering the chemicals based on 2D RDKit descriptors only. In other words, we don’t want to include “ID” and “Aff” column, which is the biological endpoint that we are interested in predicting. ###
myvars <- names(csv_data) %in% c("ID","Aff")
desc <- csv_data[!myvars]### We will then set the first column as row names. ###
rownames(desc) <- csv_data[,1]
### We will then normalize columns. Note that if you are using binary data for fingerprints like MACCS or ECFP, you don’t need to do this step. ###
matTrans <- scale(desc)
### We will then compute the distance matrix for the previously scaled dataset. The default for dist function is Euclidean distance. Based on the dataset and the goal of your project, you can tweak the distance functions as you see fit. ###
d <- dist(matTrans)
### Now, we will apply unsupervised hierarchical clustering with ward linkage to our distance matrix. ###
tupgma <- upgma(d, method="ward.D2")
### At this point, we already have a network structure of your chemical. We will then export the edges from our chemical network and prepare them for Gephi visualizations. ###
write.csv2(tupgma$edge, file = "edges.csv")
I hope it has been quite easy and straightforward to follow. You should certainly familiarize yourself with distance methods and clustering algorithms so you can apply appropriate techniques based on your dataset. In this post, I am focusing on how we can generate the chemical network visualizations so I won’t be going over those details in this blogpost.
Once you get to that point, you can prepare the input file for gephi network. Open Excel go to “Data” tab and click “From Text/CSV”. Then, load the “edges.csv” that you previously generated (screenshot shown below).

Now, you will copy the two V1 and V2 columns into a new Microsoft excel file and name them “Source” and “Target” respectively. Then add two more columns named “Type” and “Weight”. Fill “Type” Column to the end of the data with “undirected”, and “Weight” with “1” (screenshot shown below). Save it as “gephi_edges.csv” file.

Now you have successfully prepped the edges file (“gephi_edges.csv”) required for generating the gephi network visualization. You still need to prep your nodes file but it’s much more straightforward.
Make a copy of the original file “2Drdkit_chembl364.csv” that you used for conducting unsupervised hierarchical clustering and name it “gephi_nodes.csv”. It should have a list of all the chemicals and their attributes that you might want to get incorporated in the network.
Insert a new column called “Id” at the very first column. This is specific to our network and different from the ids of compounds. These are node IDs, and Gephi will use that first column to connect the nodes, form edges and eventually a network. Then, fill that very first “Id” column with number series starting from 1 and incrementing with 1. For clarification purposes, we will rename the original “ID” column containing chembl ids to “ChEMBL_IDs” (screenshot shown for prepped nodes file).
You can remove some columns/attributes that you aren’t interested in incorporating into the network. Every column in that file can be used to color or change the size of the nodes in the gephi chemical network.

Now you have both nodes (“gephi_nodes.csv”) and edges (“gephi_edges.csv”) files ready for importing into Gephi.
Next, open up Gephi and create a new project. In the tab “Data Laboratory”, click “Import Spreadsheet” and upload your “gephi_nodes.csv” file. If you do everything correctly, it should detect that it’s a nodes file.

Click “Next” and “Finish” in the next two steps. Make sure to select “Append to existing workspace” when prompted. It may indicate some errors that are shown as SEVERE for some values or columns. In our example case, they come from columns I am not that interested in fixing, so I will ignore them. You can use your best judgement based on the datasets and the nature of the project that you are working on. Then, click “OK”. Now, you have imported your nodes file.

Let’s import the edges file next. Click “Import Spreadsheet” again and select your “gephi_edges.csv” file. If correctly done, it will automatically detect that it’s the Edges table. Click “Next” and “Finish” in the next two steps. In the following step, make sure to choose “Append to existing workspace” and click “OK” (screenshot shown below).

Then, go to overview tab. This is where you will manipulate and create network visualizations of your chemicals and how they look like. There are several default algorithms already installed (if not, you can download and install them) that you can apply to your network. Once you get to that point, it becomes your playground for creativity. You can use multiple layouts and adjust a lot of parameters based on your project needs. I usually like Multigravity ForceAtlas 2 and Yifan Hus layout algorithms for my chemical networks. Contraction and expansion are quite useful as well. I have attached several tutorial GIFs below for your convenience.







Below are three sample output files generated from this tutorial.



Once you get into Gephi, there are already some tutorials out there that specifically focus on what type of visualizations you want, what you can do, etc. As far as I know, there hasn’t been a tutorial on how to actually prepare input chemical data for Gephi and I wanted to provide that missing link for Cheminformatics practitioners.
Please cite this blog if you use this technique for your research articles.
I will probably make a tutorial video on the entire process with narration and put it on youtube in the near future for those who prefer watching videos. I hope it has been helpful. Cheers!